
Translate this page into:
Cerebrotendinous Xanthomatosis – Case Series Highlighting the Role of Imaging in Diagnosis

*Corresponding author: Birma Ram, Department of Radiodiagnosis, Command Hospital, Kolkata, West Bengal, India. innovativehooda@yahoo.co.in
-
Received: ,
Accepted: ,
How to cite this article: Ram B, Gopinath M, Sharma P, Maurya V, Mukherjee G. Cerebrotendinous Xanthomatosis – Case Series Highlighting the Role of Imaging in Diagnosis. Indian J Musculoskelet Radiol 2020;2(2):115-9.
Abstract
Cerebrotendinous xanthomatosis is a rare genetic disease with an autosomal recessive inheritance. It is characterized by deposition of cholesterol and cholestanol in various tissues of the body. Imaging features are characteristic and help to clinch the diagnosis. Early diagnosis is extremely important as timely treatment with chenodeoxycholic acid and HMG-CoA reductase inhibitors can halt the further progression of the disease.
Keywords
Cerebrotendinous xanthomatosis
Ultrasound
Dentate nuclei
Tendon xanthoma
Magnetic resonance imaging

INTRODUCTION
Cerebrotendinous xanthomatosis (CTX) is an uncommon genetic disorder with an autosomal recessive inheritance.[1] It is a lipid storage disorder caused by mutations in the CYP27A1 gene, located on chromosome 2q33 which is responsible for encoding sterol-27 hydroxylase, prime enzyme for bile acid biosynthesis.[2] There is genetic defect in bile acid synthesis along with characteristically elevated blood cholestanol level, whereas the serum cholesterol can be within normal range.[3] The etiopathogenesis of the disease is the increased deposition of cholesterol and cholestanol in various tissues, predominantly in the central nervous system, tendons, and less commonly in peripheral nerves, lungs, liver, and kidneys.[3] The characteristics manifestation of the disease includes childhood diarrhea, juvenile cataract, mental retardation, and cerebellar ataxia along with tendon xanthomas. Early diagnosis is extremely important because medical treatment with chenodeoxycholic acid, if started early, can prevent the further deterioration of the disease.[4,5] We present two cases of adolescent patients with HPE proven diagnosis of CTX where imaging played a pivotal role in diagnosis.
CASE-1
A 24-year-old male presented with history of multiple falls, difficulty in carrying out complex tasks and seizures for the past 4 years. He was operated on for cataract in both eyes at the age of 8 years. At presentation to our institute, higher mental functions were normal. Power was normal in all four limbs. On clinical examination, patient had ataxic gait and intention tremors.
Mild hypertonia was observed. Hyperreflexia was noted and plantar reflux was up going bilaterally. Lobulated soft-tissue swellings were noted in the posterior aspects of the ankles and also in the anterior aspect of the knees.
The hematological and biochemical investigations were within normal range. Nerve conduction studies were normal. USG abdomen revealed bilateral non-obstructive renal calculi.
NCCT head revealed diffuse cerebellar atrophy with nearly symmetrical hypodensities in the dentate nuclei and the cerebellar white matter [Figure 1].
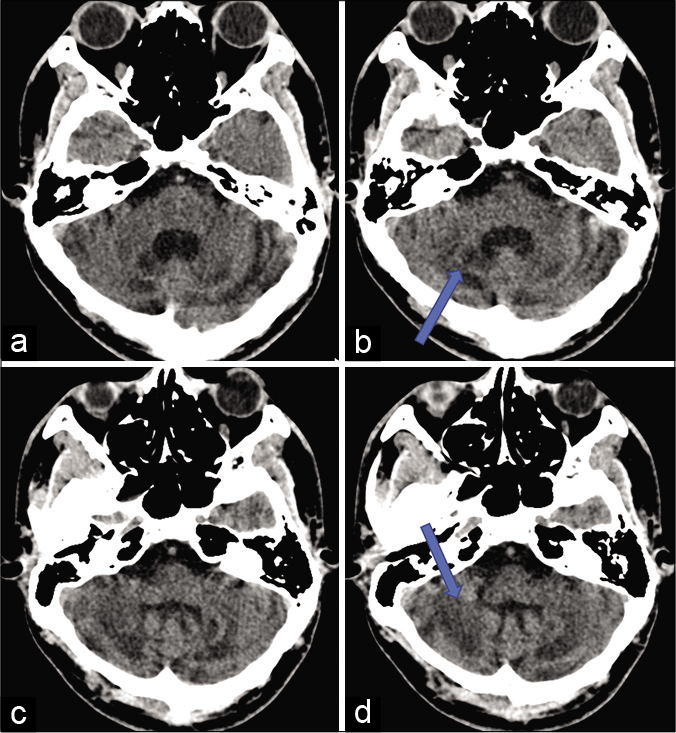
- NCCT head axial section showing diffuse cerebellar atrophy with nearly symmetrical hypodensities (shown by arrow) in the dentate nuclei (a and b) and the cerebellar white matter (c and d).
USG of the ankle and the knee revealed tendon swelling with loss of fibrillary pattern in the tendo-achilles and the patellar tendons (images not provided). T1W and T2W sagittal images of the right ankle [Figure 2a and b] and left knee [Figure 2c and d] demonstrating tendon xanthomas showing intermediate signal intensity on both sequences.

- Sagittal MRI T1 and T2 images of the ankle (a and b) and the knee (c and d) showing tendon enlargement with tendon xanthomas showing intermediate signal intensity on both sequences in the tendo Achilles and the patellar tendons.
MRI brain had shown diffuse cerebellar atrophy [Figure 3a] and bilaterally symmetrical hyperintensities in dentate nuclei and the cerebellar white matter on T2/FLAIR sequences [Figure 3b, c and e]. Lesions demonstrate hypointense rims on T2WI suggestive of xanthomas [arrow in Figure 3b]. On T1WI these lesions appear hypointense [Figure 3f]. There was no blooming seen on GRE sequences. No restriction of diffusion was noted. T2/FLAIR hyperintensities were also noted in white matter and the cerebral peduncle [Figure 3d].

- MRI, T2WI revealing diffuse cerebellar atrophy (a) with bilaterally symmetrical hyperintensities in dentate nuclei and the cerebellar white matter (b and c). Lesions demonstrate hypointense rims suggestive of xanthoma (arrow in b). Hyperintense signal intensities were also noted in white matter and the cerebral peduncle (d). FLAIR axial section showing hyperintensities in dentate nuclei and the cerebellar white matter (e). On T1WI these lesions appear hypointense (f).
Diagnosis was confirmed on histopathological evaluation of biopsy taken from the tendo achilles, which demonstrated cholesterol crystals, collection of bland histiocytes with pyknotic nucleus, as well as occasional foamy histiocytes and mild lympho nuclear infiltrate [Figure 4]. There were foci of increased fibrosis. This was reported as a tendon xanthoma.
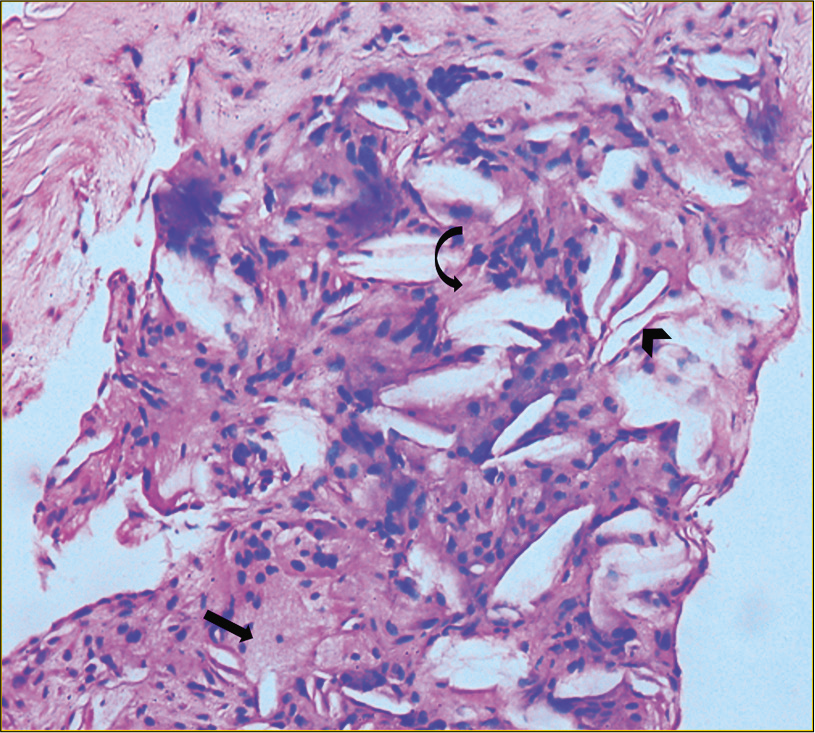
- Biopsy taken from the tendo Achilles demonstrates cholesterol crystals (arrowhead), collection of bland histiocytes with pyknotic nucleus as well as occasional foamy histiocytes (black arrow) and mild lympho nuclear infiltrate. There are foci of increased fibrosis (curved arrow).
CASE-2
A 20-year-old woman presented with history of single episode of unprovoked tonic clonic seizure. She had undergone ophthalmic surgery for bilateral cataract in her early childhood. She had a history of multiple episodes of diarrhea in childhood. Her poor scholastic performance was reported by her parents. Achilles tendon was enlarged bilaterally on clinical examination. Her neurological examination was unremarkable. There was no history of dyskinesia or ataxia at presentation. The hematological and biochemical investigations were within normal range.
Bilateral soft-tissue opacities without calcification are noted in the posterior aspect of the ankle on the plain lateral radiographs of both the ankles [Figure 5a], there was mild obliteration of Kager’s pre achilles fat pad. On ultrasound of the ankle, thickening of achilles tendon with loss of the normal fibrillary architecture and smooth, symmetric, hypoechoic infiltration was noted bilaterally [Figure 5b]. MRI of both ankles revealed enlarged achilles tendon with hypointensities on T1W and T2W sequences [Figure 5c and d].
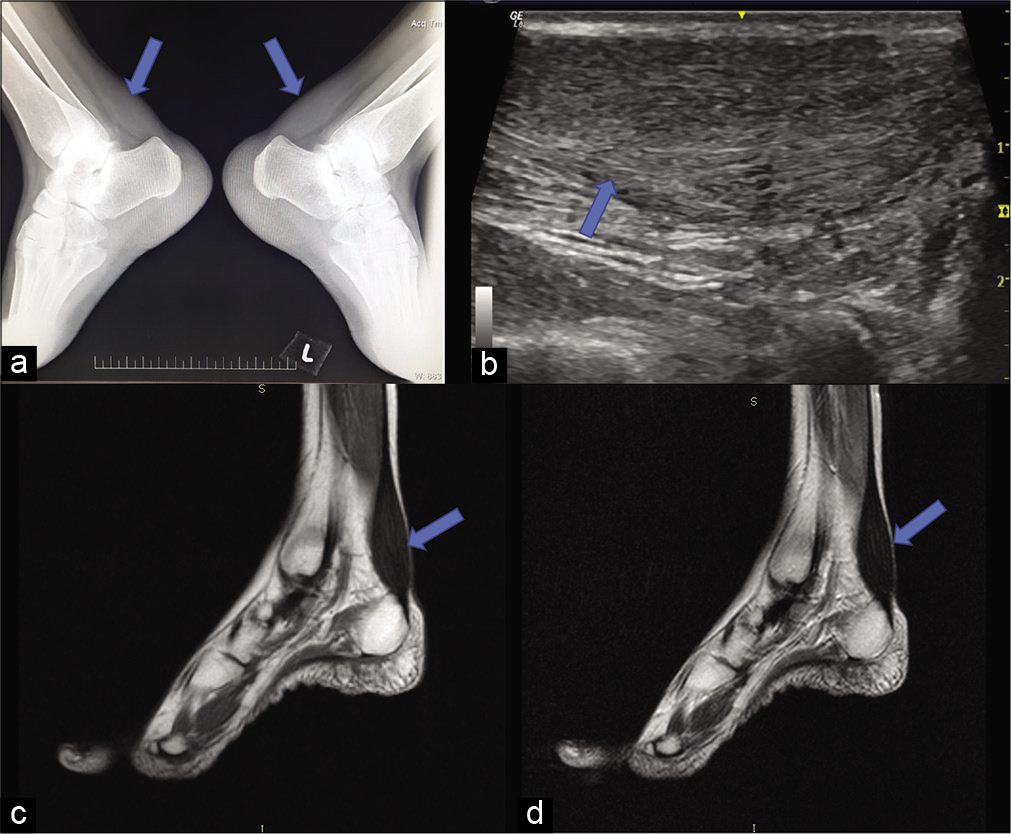
- Imaging of the ankles showing enlarged Achilles tendon with obliteration of the Kager’s fat pad. Plain lateral radiographs of both ankle (a) obliteration of retro-calcaneal fat pad by soft-tissue density, without calcification. (b) USG sagittal image shows thickening and bilaterally symmetric hypoechoic infiltration and loss of normal fibrillary architecture. (c and d) MRI, T1 and T2 sagittal image of ankle showing enlarged tendon (arrows).
MRI brain revealed mild cerebellar atrophy [Figure 6a] and bilaterally symmetrical hyperintensities involving the dentate nuclei and the deep cerebellar white matter on T2 and FLAIR sequences [Figure 6b and c] and hypointensities on T1 in corresponding areas [Figure 6d]. The basal ganglia, thalamus, and supratentorial brain parenchyma were unremarkable. NCCT head was unremarkable. The diagnosis was made on clinicoradiological correlation and HPE was not done.

- MRI brain of 20-year-old female, (a) T2 sagittal section showing mild cerebellar atrophy. (b and c) T2 and FLAIR axial section showing bilaterally symmetrical hyperintensities involving the dentate nuclei and the deep cerebellar white matter (arrow), which appears iso to hypointense on T1 (arrow in d).
DISCUSSION
CTX is a rare autosomal-recessive, inborn metabolic disorder of bile acid synthesis. The average age of presentation is 35 years with and a diagnostic delay of 16 years.[1] Mutations in the CYP27A1 gene located on chromosome 2q33-qter are implicated, leading to absence of mitochondrial enzyme sterol 27 hydroxylase, which normally catalyzes the oxidation of cholesterol into bile acids.[1,2] There is deficiency in bile acid synthesis along with increased deposition of cholesterol and cholestanol in multiple tissues.[2] Diverse manifestations of the disease can be divided into neurological and non- neurological symptoms.[3] The neurological manifestations includes poor scholastic performance, dementia, mental retardation, cerebellar ataxia, dystonia, spinal cord paresis, and psychiatric symptoms.[3,4] The other manifestations of the disease can be diarrhea juvenile cataracts in infancy or early childhood and tendon xanthomas.[4,5]
The predominantly affected tendons are Achilles and the quadriceps tendon and less commonly triceps and finger extensor tendons. Our patients had history of childhood diarrhea, cataracts, and Achilles tendon xanthomas on imaging.
Both our patients had history of childhood cataracts and operated in early childhood. Both patients had history of poor academic performance at school; however, higher mental functions were within normal limits without any obvious history of psychiatric illness. No signs of premature atherosclerosis were seen. The symptomatology and examination findings demonstrated all the signs of cerebellar involvement along with multiple tendon xanthomas.
Hokezu et al., in a study including eight patients with CTX, reported only mild or equivocal cerebral atrophy even in cases with severe mental deterioration.[4] Our patient showed evidence of only cerebellar atrophy with white matter hypodensities in the cerebellum. No evidence of cerebral atrophy was noted.[4]
Diffuse or focal FLAIR hyperintense lesions due to demyelination in the supratentorial white matter cerebral peduncle, globus pallidus has been reported.[6,7] Or patient demonstrated mild white matter changes and lesions of the cerebral peduncle.
Most frequent imaging findings reported in the literature include cerebellar atrophy along with bilateral symmetric T2 hyperintense lesions in the dentate nuclei and the cerebellar white matter along with cerebellar atrophy. A characteristic T2 hypointense rim has been described.[8] A few hypointense foci suggestive of hemorrhage or calcification can be noted within the hyperintensities involving the dentate nuclei and the cerebellar white matter.[7] Our patients had all these characteristic findings. As the disease advances, demyelinated areas appear hyperintense on T2WI with peripheral rim of hypointensity which are caused by the xanthomas.[7,8]
Atrophy of the cord, brainstem, and corpus callosum, including lesions of the spinal lateral and dorsal columns are uncommon.[8] Our patients did not reveal any focal lesion of the spinal cord on MRI imaging.
Due to deposition of free cholesterol and cholestanol, tendon xanthomas appear hypointense on T1- and T2-weighted sequences. Unaffected tendon normally appears hyperintense on T1WI due to fat signal intensity of triglycerides and fatty acids.[8] The extent of intra-tendinous lesions can be assessed on high-resolution USG as well as on MRI. Progression of the disease can be monitored using high-resolution USG of the Achilles tendon. The reduction in anteroposterior diameter of the tendons suggest the better response of the treatment.[7] In our experience, USG is preferable for the follow-up as it is easily available, has no risk of radiation and equal efficiency.
Histopathological examination of the tendon masses shows collection of xanthoma cells with multiple, dispersed lipid crystal clefts.[8] Biopsy taken from the tendo Achilles in our patient demonstrated cholesterol crystals, collection of bland histiocytes with pyknotic nucleus as well as occasional foamy histiocytes and mild lympho nuclear infiltrate. There were foci of increased fibrosis.
Other lipid storage disorders such as homozygous familial hypercholesterolemia familial dysbetalipoproteinemia and sitosterolemia are common differential diagnosis of CTX. The characteristics finding such as history of juvenile cataracts and progressive neurologic dysfunction distinguishes CTX from all of these disorders. All these lipid storage disorder can present with xanthomas and cardiovascular diseases. The MarinescoSjogren syndrome, an autosomal recessive disorder closely mimics CTX, clinically. The diagnostic triad of congenital cataract, cerebellar ataxia and mental retardation are the typical manifestations. It can be differentiated from CTX by the absence of tendon xanthomas and presence of scoliosis and small bones of the hand and feet.[9] Precise diagnosis and differentiation between these two entities are important as CTX is a medically treatable condition.[9] Hyperintensity of dentate nucleus on T2/ FLAIR WI of brain MRI can be seen in metronidazole toxicity and decompensated Maple syrup urine disease which can be differentiated from CTX by clinical evaluation including history and presence of acute encephalopathy.[10]
The final diagnosis can be confirmed based on the clinical presentation, the presence of raised serum cholestanol levels and the characteristic imaging findings. Biopsy, though it is a gold standard, but not always required in the presence of all of these imaging and clinical features.
These patients can be managed conservatively with chenodeoxycholic acid and HMG-CoA reductase inhibitors. The medical treatment must be started at early stage of the disease to prevent further deterioration of the disease.[8]
Summary
Early diagnosis of CTX using multimodality approach has paramount importance. The medical management with HMGCoA reductase inhibitors and chenodeoxycholic acid generally improves the neurological and non-neurological manifestation of the disease and contributes to a better prognosis. These patients respond to treatment gradually over a period of time and clinical improvement takes time. Therefore, long-term follow-up with periodic evaluation and imaging of brain, ankle, and knee is advised in diagnosed cases of CTX.
Declaration of patient consent
Patient’s consent not required as patients identity is not disclosed or compromised.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Cerebrotendinous xanthomatosis in Spain: Clinical, prognostic and genetic survey. Eur J Neurol. 2011;18:1203-11.
- [CrossRef] [PubMed] [Google Scholar]
- Mutations in the bile acid biosynthetic enzyme sterol 27-hydroxylase underlie cerebrotendinous xanthomatosis. J Biochem. 1991;266:7779-83.
- [Google Scholar]
- Cerebrotendinous xanthomatosis: Clinical and biochemical evaluation of eight patients and review of literature. J Neurol Sci. 1991;102:225-32.
- [CrossRef] [Google Scholar]
- Cerebrotendinous xanthomatosis: Cranial CT and MRI studies in eight patients. Neuroradiology. 1992;34:308-12.
- [CrossRef] [PubMed] [Google Scholar]
- Juvenile cataract associated with chronic diarrhea in pediatric cerebrotendinous xanthomatosis. Am J Ophthal. 1991;112:606-7.
- [CrossRef] [Google Scholar]
- Cerebrotendinous xanthomatosis: Neuroimaging findings in two siblings from an Indian family. Neurol India. 2003;51:401-3.
- [Google Scholar]
- Cerebrotendinous xanthomatosis (CTX): A treatable lipid storage disease. Pediatr Endocrinol Rev. 2009;7:6-11.
- [Google Scholar]
- Cerebrotendinous xanthomatosis: The spectrum of imaging findings and the correlation with neuropathic findings. Radiology. 2000;217:869-76.
- [CrossRef] [PubMed] [Google Scholar]
- Cerebrotendinous xanthomatosis without tendon xanthomas mimicking Marinesco-Sjogren syndrome: A case report. J Neurol Neurosurg Psychiatry. 1996;60:582-5.
- [CrossRef] [PubMed] [Google Scholar]
- Cerebrotendinous xanthomatosis: A comprehensive review of pathogenesis, clinical manifestations, diagnosis, and management. Orphanet J Rare Dis. 2014;9:179.
- [CrossRef] [PubMed] [Google Scholar]




