
Translate this page into:
Osteolytic variant of polyneuropathy, organomegaly, endocrinopathy, M-protein, skin changes syndrome in a young male – An elusive cause of polyneuropathy


*Corresponding author: Priyadharshini Bargunam, Department of Pathology, Vardhman Mahavir and Safdarjung Hospital, New Delhi, India. priyasweetygunam@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Saifi AS, Singh U, Jalan D, Bargunam P. Osteolytic variant of polyneuropathy, organomegaly, endocrinopathy, M-protein, skin changes syndrome in a young male – An elusive cause of polyneuropathy. Indian J Musculoskelet Radiol. doi: 10.25259/IJMSR_69_2024
Abstract
Polyneuropathy, organomegaly, endocrinopathy, M-protein, skin changes (POEMS) syndrome is a rare paraproteinemic paraneoplastic syndrome encountered in the sixth decade with underlying monoclonal plasma cell proliferation and polyneuropathy. The osseous lesions seen are most commonly osteosclerotic. We present a rare case of POEMS syndrome in a 24-year-old patient with osteolytic lesions and fulfillment of the mandatory (monoclonal plasma cell proliferation and polyneuropathy), major (Castleman’s disease on lymph node biopsy), and minor (organomegaly, papilledema) criteria. This case report underlines that the diagnosis of POEMS syndrome should not be discounted in young patients and in the setting of lytic osseous lesions when other diagnostic criteria have been met to mitigate irreversible organ damage.
Keywords
Castleman’s disease
Chronic inflammatory demyelinating polyradiculoneuropathy
Plasmacytoma
POEMS syndrome
Young adult

INTRODUCTION
Plasma cell dyscrasias form a heterogeneous group of age-related monoclonal plasma cell proliferative disorders. The spectrum comprises solitary plasmacytoma (intraosseous or extramedullary), monoclonal gammopathy of undetermined significance, smoldering multiple myeloma (MM), MM, Waldenstrom macroglobulinemia, and polyneuropathy, organomegaly, endocrinopathy, M-protein, skin changes (POEMS) syndrome. POEMS syndrome is a rare paraproteinemic paraneoplastic syndrome characterized by the presence of both the mandatory criteria of monoclonal plasma cell proliferation and polyneuropathy. There is multi-organ involvement with a median age of onset in the 6th decade and a slight male preponderance.[1] The initial presenting symptom is commonly that of neuropathy.[2,3] The diagnosis goes above and beyond the acronym and is established by fulfilling both mandatory criteria with at least one feature each from the major and minor criteria as discussed in Table 1.[4] The associated bone lesions are characteristically osteosclerotic in the majority of cases. We discuss a rare case of POEMS syndrome in a young male patient who presented with polyneuropathy and osteolytic lesions.
| Criteria | Feature |
|---|---|
| Mandatory (both criteria required) |
|
| Major (any one of the three required) |
|
| Minor (any one of the six required) |
|
IMWG: International myeloma working group, VEGF: Vascular endothelial growth factor, POEMS: Polyneuropathy, organomegaly, endocrinopathy, M-protein, skin changes
CASE REPORT
A 24-year-old male presented with a 3-month-long history of gradually progressive weakness in bilateral lower limbs and a resultant wheelchair-bound state. Physical examination revealed bilateral cervical lymphadenopathy. Neurological examination revealed 0/5 power in plantar flexion and 1/5 power in flexion at the hip with areflexia, sensory loss, and painful sensations in bilateral lower limbs. Hyporeflexia was present in bilateral upper limbs with intact (5/5) power. Nerve conduction studies revealed a chronic inflammatory demyelinating polyneuropathy (CIDP) pattern. Fundus examination revealed bilateral papilledema. Cerebrospinal fluid analysis revealed albuminocytological dissociation with 2 cells/mm3 and protein of 260 mg/dL (N: 15–60 mg/dL). Peripheral blood examination and renal function were normal. The patient underwent a contrast-enhancement magnetic resonance imaging (MRI) of the brain and spine to rule out any space-occupying lesion(s) which revealed homogeneously enhancing multifocal expansile bony lesions in the cervical and thoracic vertebral bodies and posterior elements with low signal intensity (SI) on unenhanced T1W1, high SI on T2-weighted image and thick hypointense intralesional cortical struts giving “mini-brain” appearance suggestive of plasmacytomas [Figures 1 and 2]. There was smooth cranial pachymeningeal enhancement. Brain parenchyma, spinal cord, and nerve roots were normal. A contrast-enhanced computed tomography of the chest and abdomen revealed hepatosplenomegaly and similar expansile osteolytic lesions in the right iliac bone and ipsilateral scapula [Figure 3]. Biopsy from the iliac bone revealed diffuse sheets of plasma cells with eccentric nuclei, perinuclear hof, and cartwheel chromatin along with a few immature and plasmablastic cells with high nucleus-tocytoplasmic ratio and prominent nucleoli [Figure 4a and b]. On immunohistochemistry, the tumor cells were positive for CD38 with lambda restriction [Figure 4c and d] and negative for CD45, CD20, CD138, kappa, SATB2, CD99, Pan-CK, S100, myogenin, smooth muscle actin (SMA) favoring a diagnosis of plasmacytoma.
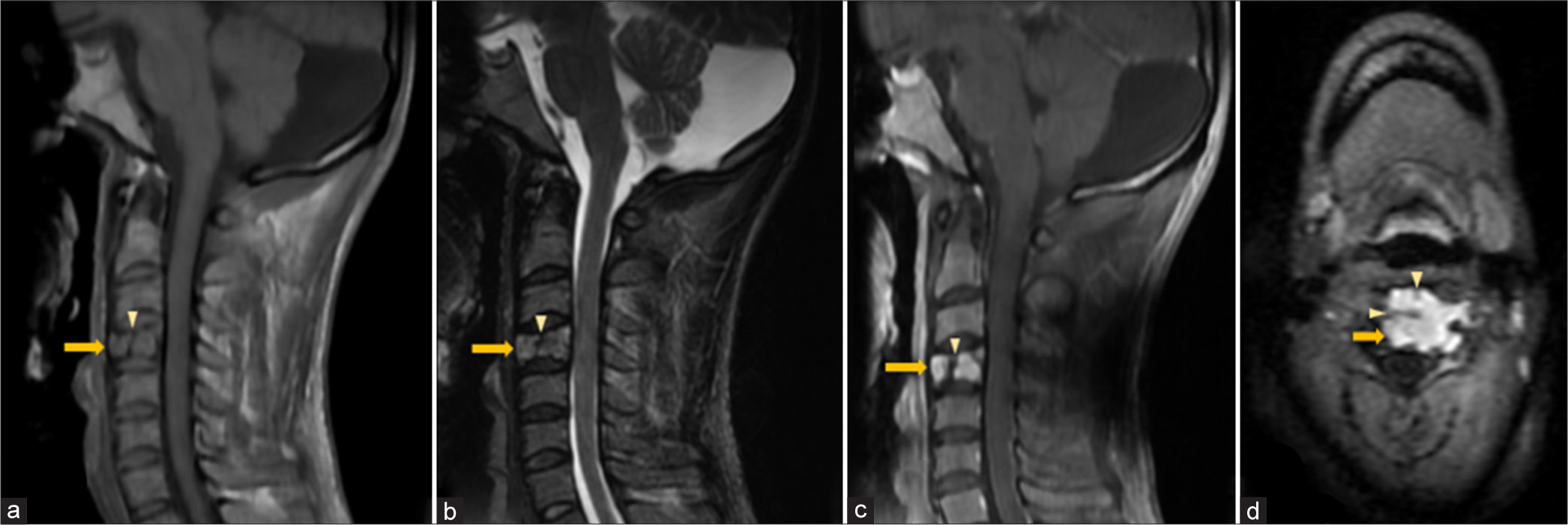
- Vertebral plasmacytoma – Sagittal (a) T1W, (b) T2W, (c) Post-contrast T1-FS and (d) axial FLAIR magnetic resonance images of cervical spine show a homogeneously enhancing geographic expansile lesion (solid arrow in a-d) in the posterior aspect of C4 vertebral body appearing hypointense on T1W and hyperintense T2W sequences with thick hypointense intralesional cortical struts (arrowhead in a-d) giving “mini-brain” appearance. FLAIR: Fluid attenuated inversion recovery, FS: Fat saturated.
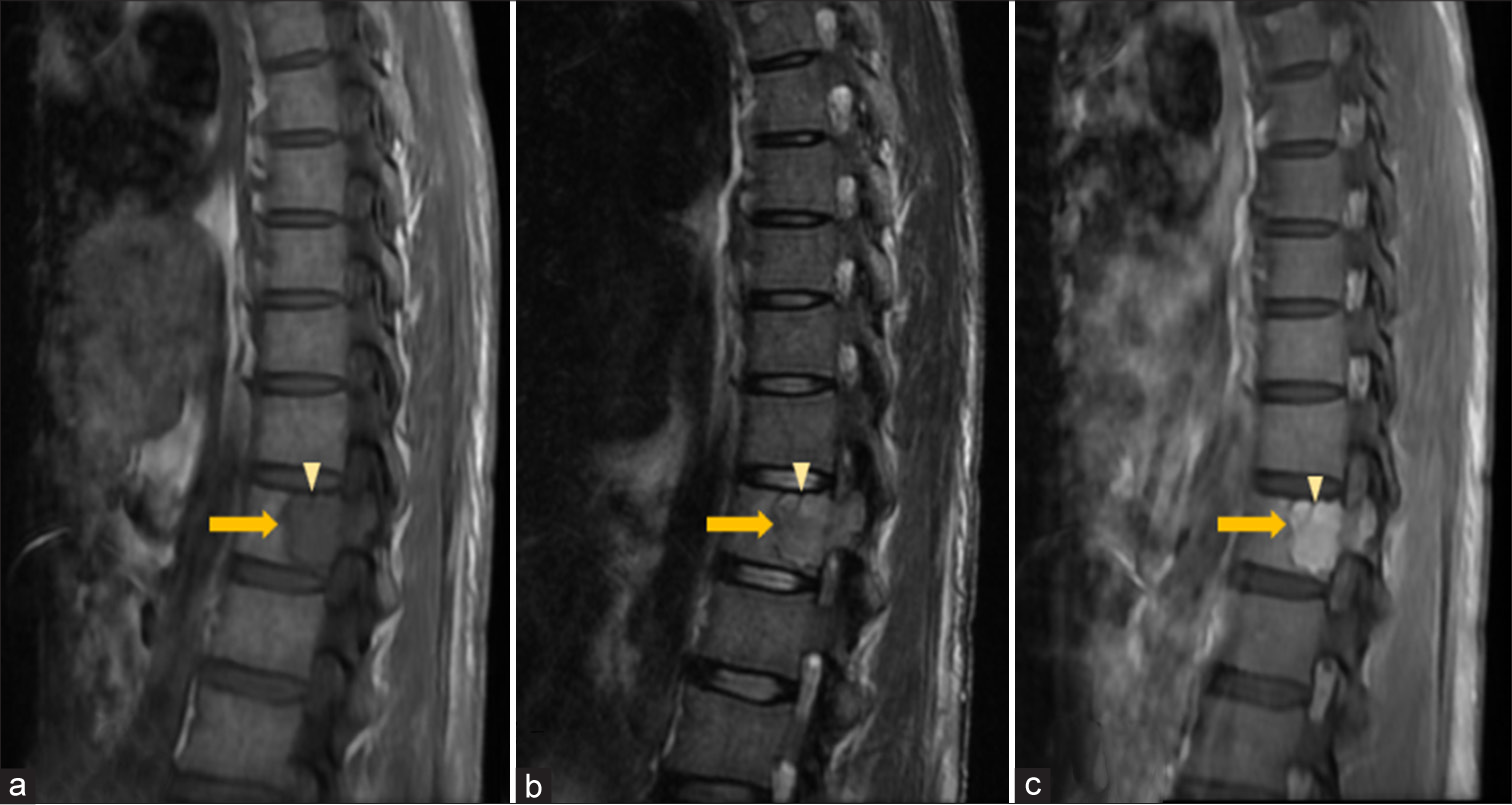
- Vertebral plasmacytoma – Sagittal (a) T1-weighted (T1W), (b) T2-weighted (T2W) and (c) Post-contrast T1-FS magnetic resonance images of thoracic spine show a homogeneously enhancing geographic expansile lesion (solid arrow in a-c) in the posterior aspect of T12 vertebral body and left pedicle appearing hypointense on T1W and hyperintense T2W sequences with a thick hypointense intralesional cortical strut (arrowhead in a-c) giving “mini-brain” appearance. FS: Fat saturated.
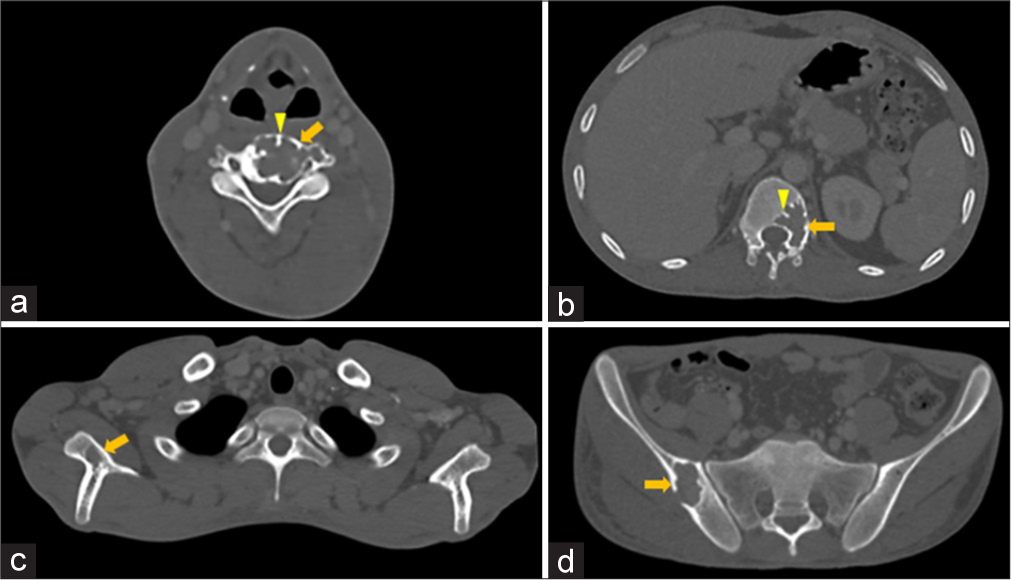
- (a-d) Osteolytic plasmacytomas – Axial computed tomography images in bone window show geographic expansile osteolytic lesions (solid arrow in a-d) with sclerotic margins and focal areas of cortical breach in (a) C4 vertebral body and left transverse process, (b) T12 vertebral body and left pedicle, (c) right scapula, and (d) right iliac blade with thick intralesional cortical struts (arrowheads in a-d) giving “mini-brain” appearance.

- Histomorphology of plasmacytomas. (a and b) Light microscopic images show diffuse sheets of plasmacytoid cells -round to oval in shape with moderate to abundant cytoplasm, eccentric nucleus, cartwheel chromatin, and a perinuclear hof on ×10 and ×40 respectively. (Hematoxylin and Eosin stain). (c) Immunohistochemistry shows strong and diffuse membranous positivity for CD38 (40X). (d) Immunohistochemistry shows lambda restriction with strong cytoplasmic positivity (10X).
Bone marrow aspirate showed <5% plasma cells (N: <10%) with normal trilineage hematopoiesis. There were abnormally elevated levels of serum lambda free light chain of 65.6 mg/L (N: 5.7–26.3 mg/L) with a faint immunoglobulin G band on serum immunofixation electrophoresis. Ultrasound-guided cervical lymph node [Figure 5] biopsy was suggestive of Castleman’s disease [Figure 6]. Vascular endothelial growth factor levels were not evaluated. A diagnosis of POEMS syndrome was made after aligning clinical features and imaging findings with laboratory investigations. However, other possible differential diagnoses on the basis of imaging alone included lymphoma (ivory vertebra appearance is more likely) and osteolytic metastases from unknown primary carcinoma (intracortical struts are unlikely) and were excluded after comprehensive corroboration with clinical and biochemical assessment. Smooth cranial pachymeningeal enhancement was presumed to be a result of lumbar puncture performed before MRI.
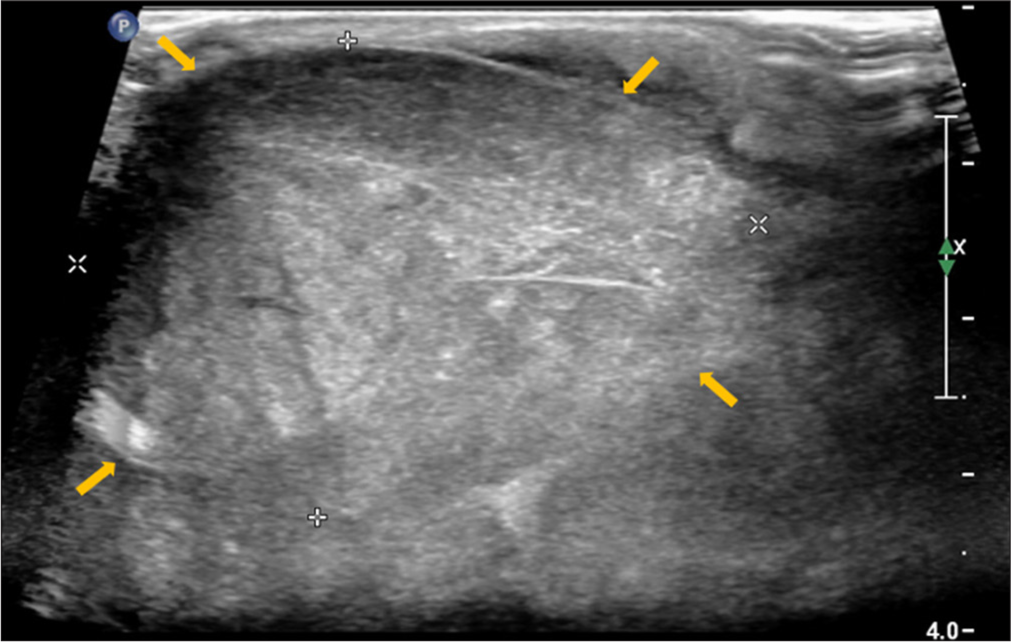
- Cervical lymph node – Short axis gray-scale sonographic image of right level III cervical lymph node showing marked enlargement and solid non-necrotic heterogeneous echotexture with effacement of normal fatty hilar morphology (yellow arrows) (biopsy showed histomorphology of Castleman’s disease).
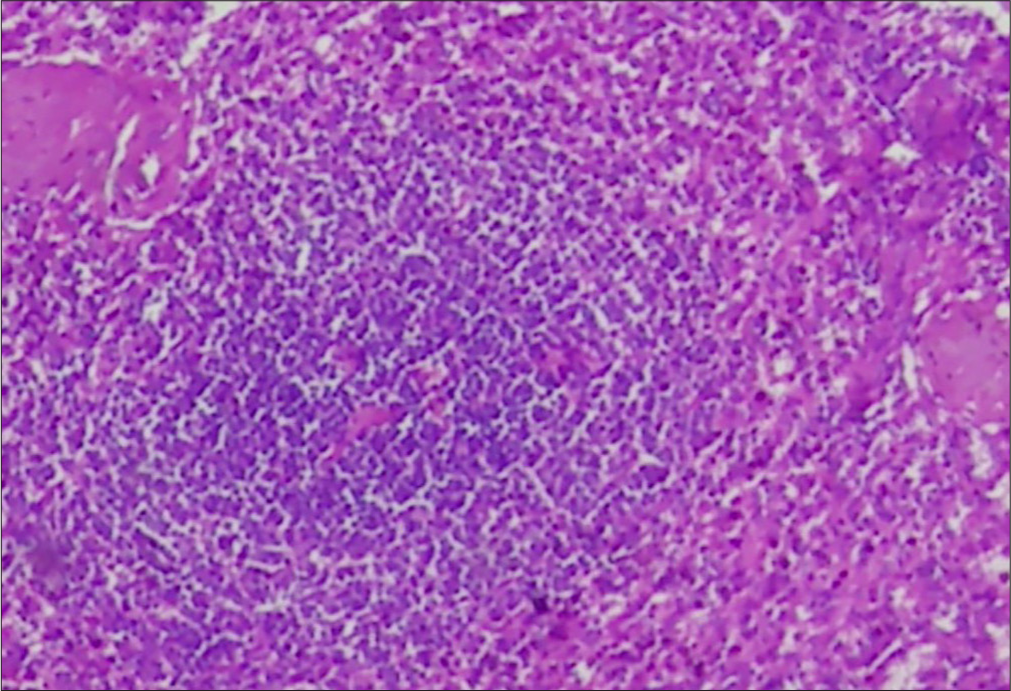
- Histopathological image of a lymph node showing features of (Hematoxylin and Eosin, 40X). Castleman’s disease -atretic follicles with hyalinized blood vessels.
DISCUSSION
POEMS syndrome is a rare paraneoplastic disorder due to monoclonal plasma cell proliferation and associated subacute onset demyelinating polyneuropathy with an estimated incidence of 0.3 cases per 1,00,000 individuals.[5] In most cases, the clonal plasma cells show lambda chain restriction. The diagnosis of POEMS syndrome is thought of in elderly male patients with polyneuropathy. Owing to its rarity, it remains an elusive diagnosis in young patients.
Our case is unique in two principal aspects – one being the presentation in a young male of 24 years age and the other being the osteolytic nature of the bone lesions.
Both mandatory criteria were fulfilled in our case with additional major (Castleman disease on lymph node biopsy) and minor criteria (organomegaly and bilateral papilledema).
Bone lesions are frequently encountered in the pelvis, thoracolumbar vertebrae, ribs, scapula, clavicle, sternum, skull, and long bones.[6] Although the most common morphology of bone lesions is sclerotic, few cases of purely osteolytic lesions in POEMS syndrome have been reported. Mayo Clinic conducted a study with 99 patients where 47% had only sclerotic lesions, 51% had mixed lytic-sclerotic lesions and only 2% had purely lytic lesions.[1] Clark et al. also reported three cases with osteolytic lesions in POEMS syndrome.[7]
One-third of patients have no widespread bone marrow involvement in POEMS syndrome with only localized bone marrow plasmacytomas and typically with <10% plasma cells on iliac crest biopsy as was seen in our case (<5%).[8]
On histomorphology, CD138 is a key marker used to identify plasma cells and is crucial for diagnosing plasma cell dyscrasias. However, a negative CD138 does not rule out a plasmacytoma.[9] A low CD138 expression indicates an immature phenotype and has a poor prognosis.[10]
Although this case lacked a CD138 expression, histomorphology along with CD38 positivity with lambda restriction ruled out a reactive plasmacytosis whereas a negative CD45 and CD20 excluded a lymphoma. Keeping in mind the unusual age of presentation, small cell osteosarcoma, Ewings as well as rhabdomyosarcomas were excluded with a negative SATB2, CD99, myogenin, PanCK, and SMA.
POEMS syndrome has seen improved overall 10-year survival rates from 55% to 79% after 2003 with the advent of treatment strategies such as autologous stem cell transplantation and immunomodulatory therapies and needs to be thought of as a differential diagnosis in similar cases.[11]
CONCLUSION
POEMS syndrome, by virtue of multi-organ involvement, has high morbidity and mortality when left untreated. The most common age range of presentation is 50–60 years and therefore remains a challenging diagnosis in young individuals. It is, thus, essential that the diagnosis of POEMS syndrome should not be discounted in young patients and in the setting of lytic osseous lesions when other diagnostic criteria have been met to mitigate irreversible organ damage.
Ethical approval:
The Institutional Review Board has waived the ethical approval for this study.
Declaration of patient consent:
The authors certify that they have obtained all appropriate patient consent.
Conflicts of interest:
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation:
The authors confirm that they have used artificial intelligence (AI)-assisted technology for assisting in the editing and grammar check of the manuscript or image creations.
Financial support and sponsorship: Nil.
References
- POEMS syndrome: Definitions and long-term outcome. Blood. 2003;101:2496-506.
- [CrossRef] [PubMed] [Google Scholar]
- Different neurological and physiological profiles in POEMS syndrome and chronic inflammatory demyelinating polyneuropathy. J Neurol Neurosurg Psychiatry. 2012;83:476-9.
- [CrossRef] [PubMed] [Google Scholar]
- Electrophysiological features of patients with POEMS syndrome. Clin Neurophysiol. 2005;116:965-8.
- [CrossRef] [PubMed] [Google Scholar]
- International myeloma working group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014;15:e538-48.
- [CrossRef] [PubMed] [Google Scholar]
- POEMS syndrome in the 21st century: A bibliometric analysis. Heliyon. 2023;9:e20612.
- [CrossRef] [PubMed] [Google Scholar]
- Osteolytic-variant POEMS syndrome: An uncommon presentation of “osteosclerotic” myeloma. Skeletal Radiol. 2017;46:817-23.
- [CrossRef] [PubMed] [Google Scholar]
- Bone marrow histopathology in POEMS syndrome: A distinctive combination of plasma cell, lymphoid, and myeloid findings in 87 patients. Blood. 2011;117:6438-44.
- [CrossRef] [PubMed] [Google Scholar]
- CD138-negative clonogenic cells are plasma cells but not B cells in some multiple myeloma patients. Leukemia. 2012;26:2135-41.
- [CrossRef] [PubMed] [Google Scholar]
- Multiple myeloma cells expressing low levels of CD138 have an immature phenotype and reduced sensitivity to lenalidomide. Int J Oncol. 2012;41:876-84.
- [CrossRef] [PubMed] [Google Scholar]
- Long-term outcome of patients with POEMS syndrome: An update of the Mayo Clinic experience. Am J Hematol. 2016;91:585-9.
- [CrossRef] [PubMed] [Google Scholar]




