
Translate this page into:
Pictorial spectrum of fibrodysplasia ossificans progressiva – Report of a rare case

*Corresponding author: Dr. Vandana Ahluwalia, MBBS, MD, DNB, MNAMS Professor and Head, Department of Radiodiagnosis, FH Medical College and Hospital, Agra, India
-
Received: ,
Accepted: ,
How to cite this article: Ahluwalia V, Qureshi AA, Ahluwalia A. Pictorial spectrum of fibrodysplasia ossificans progressiva – Report of a rare case. Indian J Musculoskelet Radiol 2023;5:64-7.
Abstract
Fibrodysplasia ossificans progressiva (FOP) is a rare debilitating inherited disorder which is characterized by great toe malformation and progressive heterotopic ossification of connective tissue in which tendons and ligaments are gradually replaced by bone. The extraskeletal heterotopic bone limits the patient’s mobility. The average age of onset is the first 2 decades of life with the current prevalence rate of 1 in 2 million cases worldwide. Thorough clinical examination, characteristic radiological findings, and genetic analysis pave a way in making an early diagnosis for better care and management of the patient with FOP.
Keywords
Fibrodysplasia ossificans progressiva
Great toe malformation
Heterotopic ossification

INTRODUCTION
Fibrodysplasia ossificans progressiva (FOP) is a rare debilitating autosomal dominant disease which is characterized by the cardinal features of congenital deformity of the big toe and progressive heterotopic ossification of connective tissues.[1,2] The average age of onset is the first two decades of life with the current prevalence rate of 1 in 2 million cases worldwide and no racial, ethnic, or geographical predilection.[3,4] Patients during the first decade of life experience acute exacerbations of sporadic episodes of painful soft-tissue swellings (flare-ups)[5] and are thus usually misdiagnosed as tumors due to their acute onset and morphological patterns.[4] At present, there is no cure for FOP; thus, the aim has been limited to preventing further ossification and to providing patients with care and rehabilitation.[4,6]
CASE REPORT
We report a case of FOP in a 32-year-old male who presented with progressive deformities and inability to bend his body from the neck down to the trunk and pelvis with a recent history of trauma. His skeletal survey and computed tomography (CT) findings with 3D reconstruction were evaluated and will be presented in a pictorial display. The case is unique due to rarity and associated right hip synovial chondromatosis which is a benign neoplasm, in which metaplastic cartilage formation occurs throughout the synovium. As of now, only a few cases of FOP with synovial chondromatosis have been reported so far.
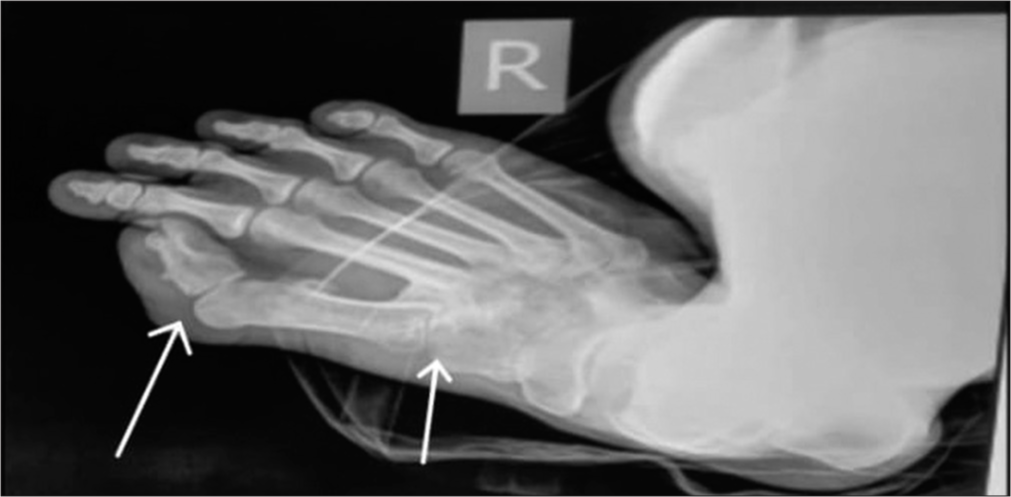
On physical examination, the patient had multiple, non-tender hard swellings in the back along with swellings in both upper and lower limbs. Radiological evaluation of the patient revealed straightening of cervical lordosis with squaring of vertebral outline. There was a fusion of vertebral bodies and neural arches, along with a thick band of soft-tissue ossification extending from the occiput to cervico-dorsal spine causing cranio-truncal fusion [Figure 1]. Frontal chest radiograph showing thick bands of heterotopic ossification resulting in bridging of the rib cage to humerus on either side or limiting lateral arm mobility [Figure 2]. Anteroposterior radiograph of the pelvis and CT scan showed broad femoral necks with thick bands of ossification extending from the left pubic bone to mid femoral shaft, along with soft-tissue ossification along the medial aspect of the thigh and left peritrochanteric region. In addition, there were multiple osteochondral bodies and associated osteoarthritic changes along the deformed and remodeled right hip with periarticular osseous ankylosis [Figure 3]. A radiograph of both hands showed microdactyly of both thumbs with fused bilateral wrist joints. The metacarpals of both hands appeared short and broad with asymmetric pronounced shortening of the 1st metacarpal bilaterally [Figure 4]. A radiograph of the right foot showed hallux valgus deformity with microdactyly due to a short broad proximal phalanx and absent distal phalanx of the great toe. There was also synostosis of the metatarsal bases with tarsal bones [Figure 5]. 3D-reconstructed CT scan in anterior, oblique, and posterior views [Figure 6] revealed the extent of heterotopic ossification from occiput to femoropelvic junction. Blood and serological studies of the patient revealed a normal hemogram, erythrocyte sedimentation rate, and serum calcium. Alkaline phosphatase, creatine phosphokinase, alanine, and routine renal function tests were all found to be within normal limits.
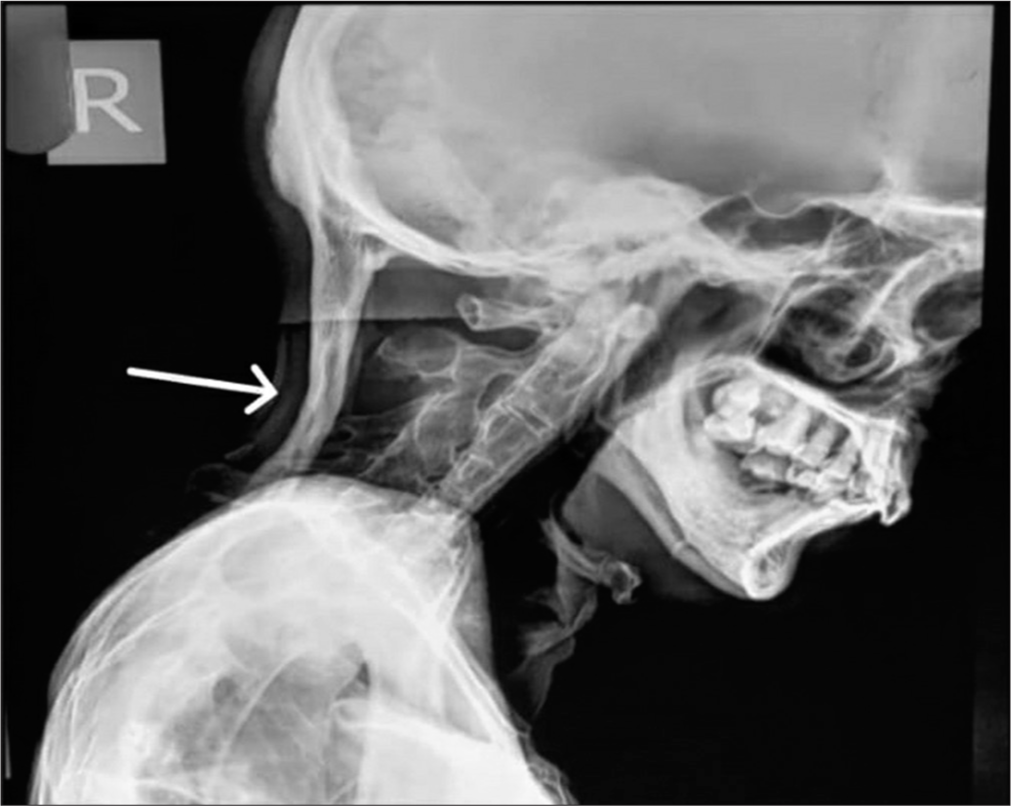
- X-ray cervical spine (lateral view) shows straightening of cervical lordosis with squaring of vertebral outline. There is fusion of vertebral bodies and neural arches. There is a thick band of soft-tissue ossification extending from occiput till cervico-dorsal spine causing cranio-truncal fusion.
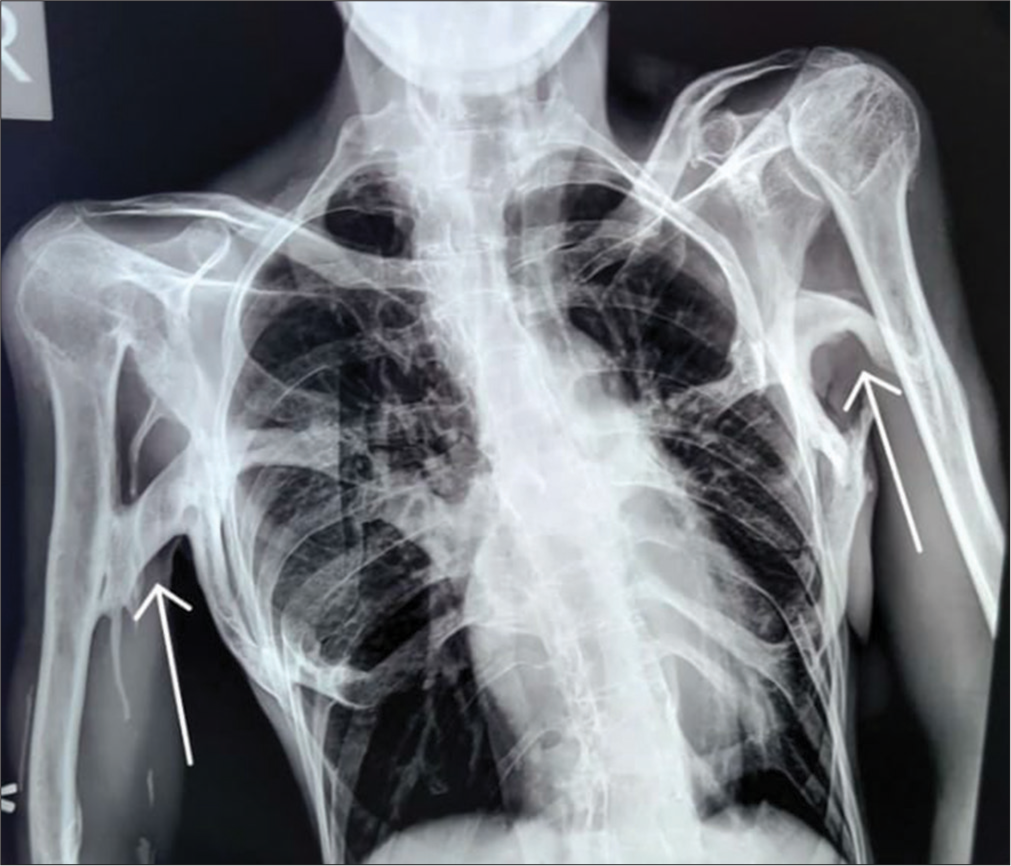
- Chest X-ray (PA view) shows thick bands of heterotopic ossification resulting in bridging of rib cage to humerus on either side and limiting lateral arm mobility.
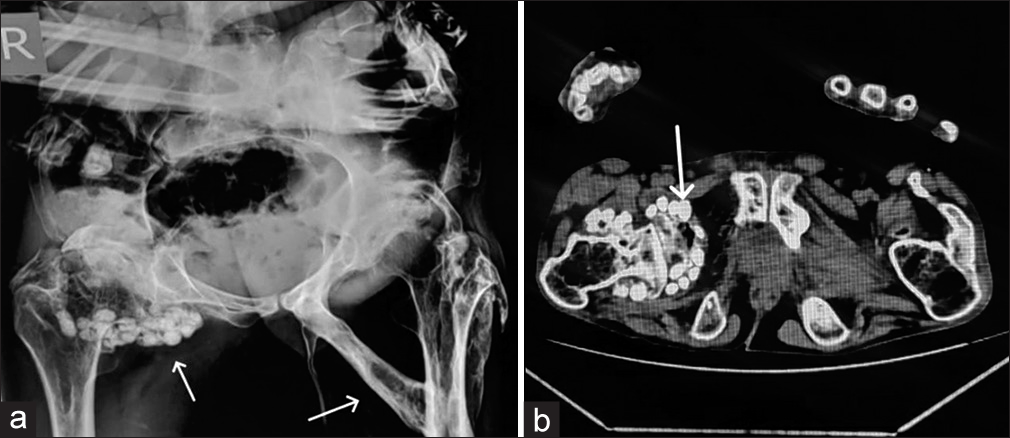
- (a and b) X-ray pelvis AP view (a) with axial computed tomography image (b) shows broad femoral necks with thick bands of ossification extending from left pubic symphysis to mid femoral shaft along with soft-tissue ossification along medial aspect of thigh and left peri-trochanteric region with multiple osteochondral bodies and associated osteoarthritic changes along deformed and remodeled right hip with periarticular osseous ankylosis.
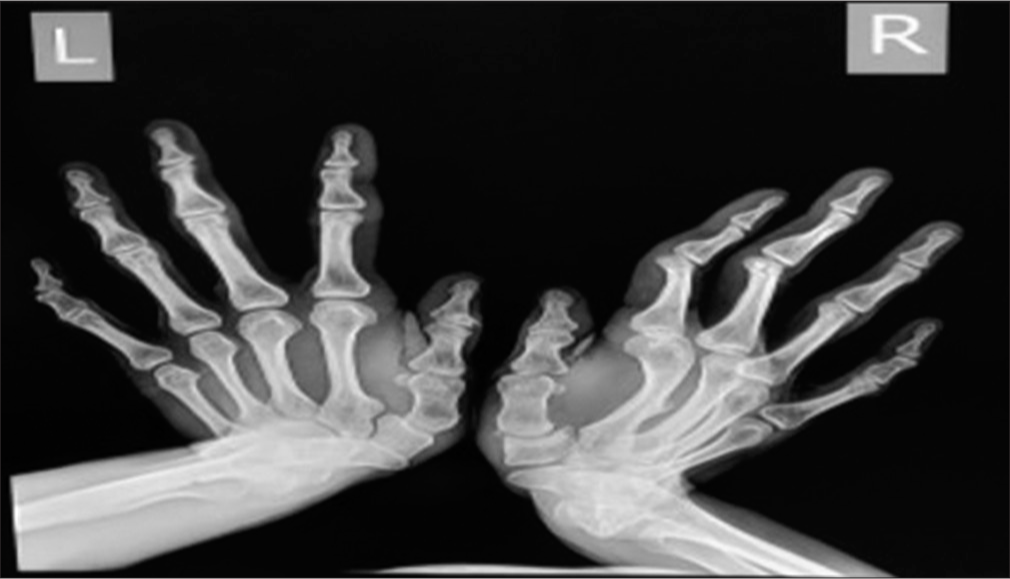
- X-ray of both hands shows microdactyly of both thumbs with fused bilateral wrist joints. The metacarpals of both hands appear short and broad with asymmetrically pronounced shortening of 1st metacarpal of bilateral hands.
- X-ray of foot shows hallux valgus deformity with microdactyly due to short broad proximal phalanx and absent distal phalanx and there is synostosis of the metatarsal bases with tarsal bones.
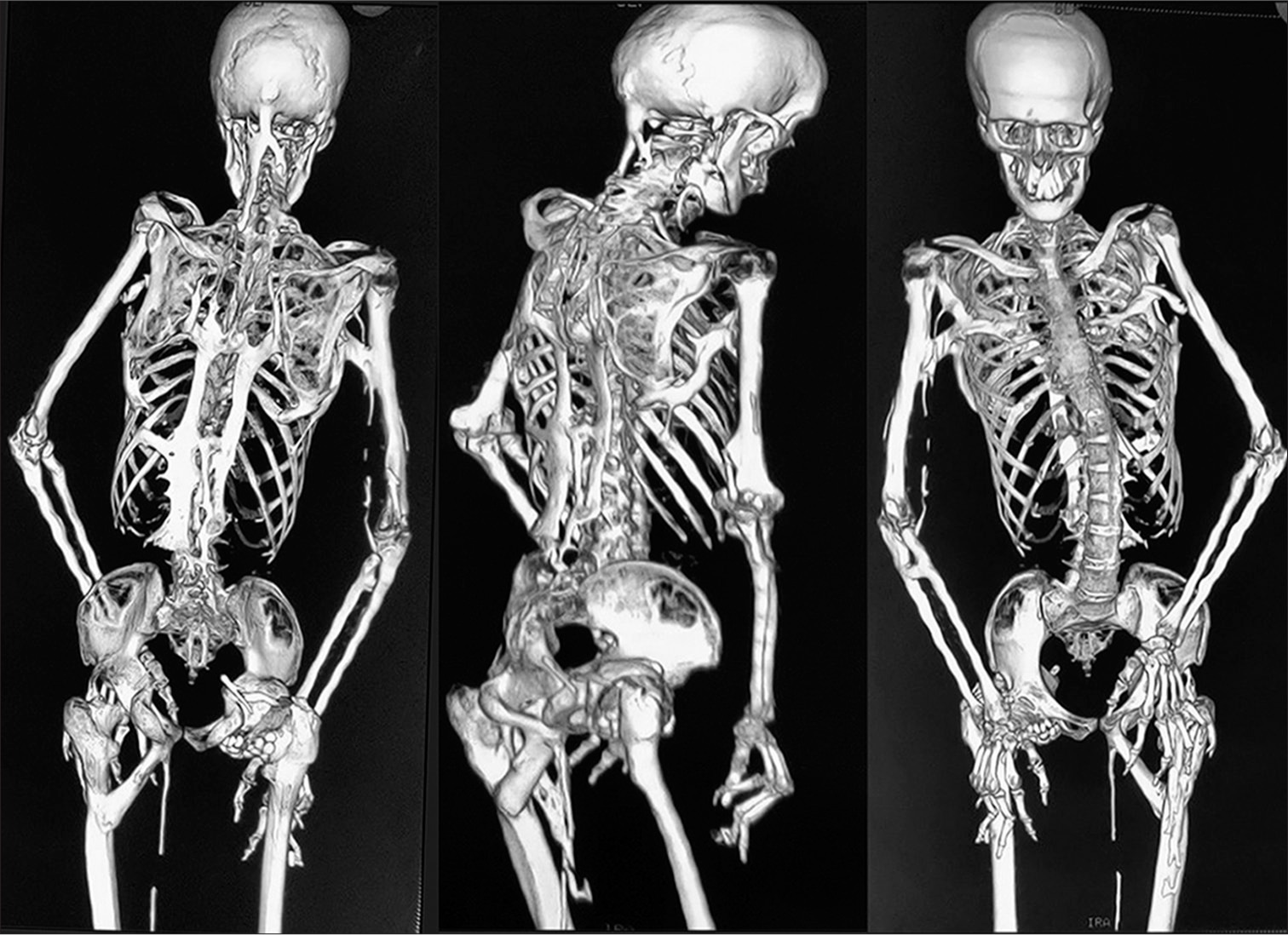
- The 3D-reconstructed computed tomography scan in posterior, oblique and anterior view reveals the extent of heterotopic ossification from occiput to femoropelvic junction.
In our patient, due to delayed presentation with fixed deformities, only symptomatic treatment was offered for the recent trauma. Routine clinical assessment and follow-up of the patient with the aim of rehabilitation and improved quality of life was provided. Further, preventive measures and lifestyle modifications with a view to reduce future injuries to the patient were also advised.
DISCUSSION
FOP is a very rare and severely disabling autosomal dominant disease having remarkably distinguishing features of heterotopic ossification characterized by the classical transformation of connective tissues into bone, and thus, the muscles, tendons, and ligaments get ossified resulting in a fixed deformity.[7] The acute flare-ups in FOP may develop spontaneously but as in our case, these are usually preceded by traumatic injuries involving soft tissues. The soft-tissue injuries can also occur due to biopsies, surgical procedures, intramuscular injections, or local anesthetics and these are also one of the leading causes of the development of flare-ups.[4,6] It has been noted that in most cases, the pattern of ossification initially involves the neck followed by the thoracic and lumbar spine with involvement of the limbs seen in later stages resulting in a fixed, immobile, and disabling condition.[8] The patients due to their inability to move or bend are thus rendered dependent and become wheelchair-bound or bedridden in the second decade of their life due to bony ankylosis of all the major axial and appendicular joints.[8]
FOP is a very rare disease and chances of misdiagnosis are very high due to the similarity with patterns of various diseases such as arthritis, rheumatic diseases, and soft-tissue sarcomas, leading to improper management of the cases.[6,9] FOP is generally diagnosed on clinical grounds based on the presence of its hallmark features of congenital anomalies of the great toes, along with progressive heterotopic ossification. Biopsies and other invasive diagnostic approaches are not recommended as they may worsen the disease process.[10]
CONCLUSION
FOP has two distinguishing cardinal features the formation of extraskeletal ossified masses that occur in a progressive and episodic manner (flare-ups) and the presence of congenital hallux valgus with microdactyly. At present, medical management is available to help in relieving the symptoms, but the best approach in a case with FOP is early diagnosis and prevention of trauma that worsens the disease. Most of the cases are misdiagnosed and mismanaged in the initial part of the disease, leading to unnecessary invasive procedures and worsening of the disease process. Diagnosis of FOP is based on clinical examination, characteristic radiological findings, and genetic analysis that help in better care and management of the patient. Early radiological diagnosis thus makes a greater impact on the correct management of the patient and promotes a holistic approach to rehabilitation.
Acknowledgment
The author acknowledges the cooperation of the patient.
Declaration of patient consent
Patient’s consent not required as patients identity is not disclosed or compromised.
Conflicts of interest
There are no conflicts of interest.
Financial support and sponsorship
Nil.
References
- Myositis ossificans of the serratus anterior as a rare complication of massage: A case report. J Med Case Rep. 2015;9:143.
- [CrossRef] [PubMed] [Google Scholar]
- Early diagnosis of fibrodysplasia ossificans progressiva. Pediatrics. 2008;121:e1295-300.
- [CrossRef] [PubMed] [Google Scholar]
- Imaging of muscle disorders in children. Pediatr Radiol. 2006;36:1005-18.
- [CrossRef] [PubMed] [Google Scholar]
- Iatrogenic harm caused by diagnostic errors in fibrodysplasia ossificans progressiva. Pediatrics. 2005;116:e654-61.
- [CrossRef] [PubMed] [Google Scholar]
- Ageand joint-specific risk of initial heterotopic ossification in patients who have fibrodysplasia ossificans progressiva. Clin Orthop Relat Res. 1994;301:243-8.
- [CrossRef] [Google Scholar]
- Challenges in the treatment of fibrodysplasia ossificans progressiva. Rheumatol Int. 2019;39:569-76.
- [CrossRef] [PubMed] [Google Scholar]
- Natural history of fibrodysplasia ossificans progressiva: Cross-sectional analysis of annotated baseline phenotypes. Orphanet J Rare Dis. 2019;14:98.
- [CrossRef] [Google Scholar]
- Fibrodysplasia ossificans progressiva: Case report. Arq Neuropsiquiatr. 2005;63:10903.
- [CrossRef] [PubMed] [Google Scholar]
- Fibrodysplasia ossificans progressiva. Best Pract Res Clin Rheumatol. 2008;22:191-205.
- [CrossRef] [PubMed] [Google Scholar]
- Fibrodysplasia ossificans progressiva. AJR Am J Roentgenol. 1982;139:93541.
- [CrossRef] [PubMed] [Google Scholar]




