
Translate this page into:
Renal tubular acidosis, osteopetrosis, and cerebral calcification – The triad of carbonic anhydrase II deficiency

*Corresponding author: Mansoor C. Abdulla, Department of General Medicine, Sultan Qaboos Hospital, Salalah, Oman. drcamans@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Saeed Al Zouabi S, Abdulla MC, Amer MS. Renal tubular acidosis, osteopetrosis, and cerebral calcification – The triad of carbonic anhydrase II deficiency. Indian J Musculoskelet Radiol. 2024;6:149-52. doi: 10.25259/IJMSR_12_2024
Abstract
A 43-year-old Omani lady was admitted with acute-onset mild weakness of all four limbs. She had a history of developmental delay and multiple fractures in the past. She had dysmorphic facies (prominent forehead, micrognathia, and misaligned teeth), low intelligence quotient (48), and grade 4/5 power in all 4 limbs. Biochemical parameters showed renal tubular acidosis (RTA). She also had osteopetrosis and cerebral calcification on evaluation. Carbonic anhydrase II deficiency (CAII deficiency) was diagnosed based on the presence of osteopetrosis, RTA, and cerebral calcification. She was managed with intravenous potassium chloride and was discharged on oral potassium chloride with sodium bicarbonate supplementation. The case highlights the importance of considering CAII deficiency in patients presenting with RTA with other features such as osteopetrosis and cerebral calcification.
Keywords
Renal tubular acidosis
Osteopetrosis
Cerebral calcification
Carbonic anhydrase II deficiency

INTRODUCTION
CAII deficiency is a genetic disorder inherited autosomal recessively and is characterized by osteopetrosis, RTA, and cerebral calcification. The presentations of patients with CAII deficiency are commonly due to manifestations related to osteopetrosis. Symptomatic hypokalemia secondary to RTA is a rare form of presentation in CAII deficiency. We present a patient with CAII deficiency who presented with weakness secondary to hypokalemia.
CASE REPORT
A 43-year-old Omani lady was admitted with acute-onset mild weakness of all four limbs. She was born as the third child of a non-consanguineous union by vaginal delivery at term without complications. Her siblings and parents were normal. She had a history of developmental delay and multiple fractures in the past. She had dysmorphic facies (prominent forehead, micrognathia, and misaligned teeth), height was 146 cm, and weight was 56 kg. She had a low intelligence quotient (48), grade 4/5 power in all 4 limbs, hypotonia, normal reflexes, and bilateral flexor plantar with normal sensory, cerebellar, bowel, and bladder functions. The rest of the systemic examination was normal.
Biochemical parameters revealed random blood sugar 4.9 mmol/L, urea 6.4 mmol per liter, creatinine 67.2 µmol/L, sodium 142 mmol/L, potassium 2.7 mmol/L, chloride 114 mmol/L, calcium 2.1 mmol/L, magnesium 0.85 mmol/L, and phosphorus 1.25 mmol/L. The liver function test was normal. Arterial blood gas analysis showed a pH of 7.29, bicarbonate 14 meq/L, and partial pressure of carbon dioxide (CO2) 27. The serum anion gap was 14. Urinary fasting pH was 7.5 (two samples), sodium 110 meq/L, chloride 102 meq/l, and potassium 24 meq/L. The urinary anion gap was positive (32). Diagnosis of renal tubular acidosis (RTA) was made since the patient had hypokalemia, normal anion gap metabolic acidosis with inappropriately high urinary potassium, and a positive urinary anion gap.
A skeletal survey showed increased bone density and thickened calvarium of the skull with bilateral basal ganglia calcifications [Figure 1a]. The spine showed increased bone density and multiple old rib fractures with callus formation [Figures 1b and c]. Pelvis and hip joints showed increased bone density, bony exostosis, and bilateral femoral neck old fractures with metallic fixation [Figure 1d]. Distal femurs and knee joints showed increased bone density with metallic fixation of proximal femoral shaft fractures [Figure 1e]. The left leg showed both bones of lower-limb fractures with metallic fixation of the distal tibia [Figure 1f]. Non-enhanced computed abdomen radiography showed bilateral renal stones, hepatosplenomegaly, and generalized increased bone density of the spine and pelvic bones, with metallic artifacts due to fixation of both femoral neck old fractures [Figure 2a-e]. Non-enhanced computed radiography of the brain showed bilateral cerebral coarse calcifications involving basal ganglia, thalami, and periventricular white matter [Figure 3a-c] and generalized increased calvarial bone density [Figure 3d].

- (a) Skeletal survey showing increased bone density and thickened calvarium of the skull with bilateral basal ganglia calcifications. (b & c) The spine showing increased bone density and multiple old rib fractures with callus formation. (d) Pelvis and hip joints showing increased bone density, bony exostosis, and bilateral femoral neck old fractures with metallic fixation. (e) Distal femurs and knee joints showing increased bone density with metallic fixation of proximal femoral shaft fractures. (f) The left leg showing both bones of lower limb fractures with metallic fixation of the distal tibia.
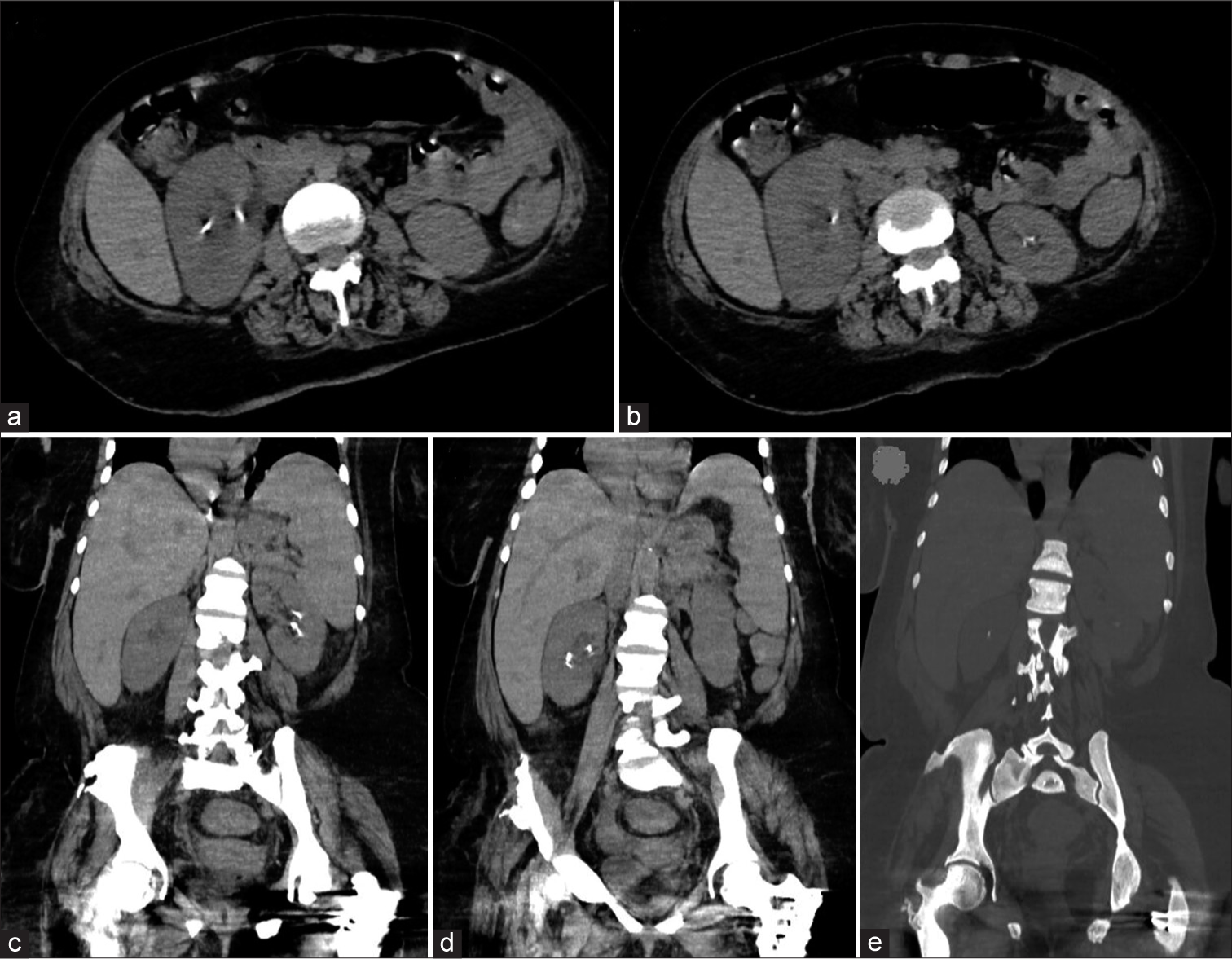
- (a-e) Non-enhanced computed radiography of the abdomen showing bilateral renal stones, hepatosplenomegaly, and generalized increased bone density of the spine and pelvic bones, with metallic artifacts due to fixation of both femoral neck old fractures.
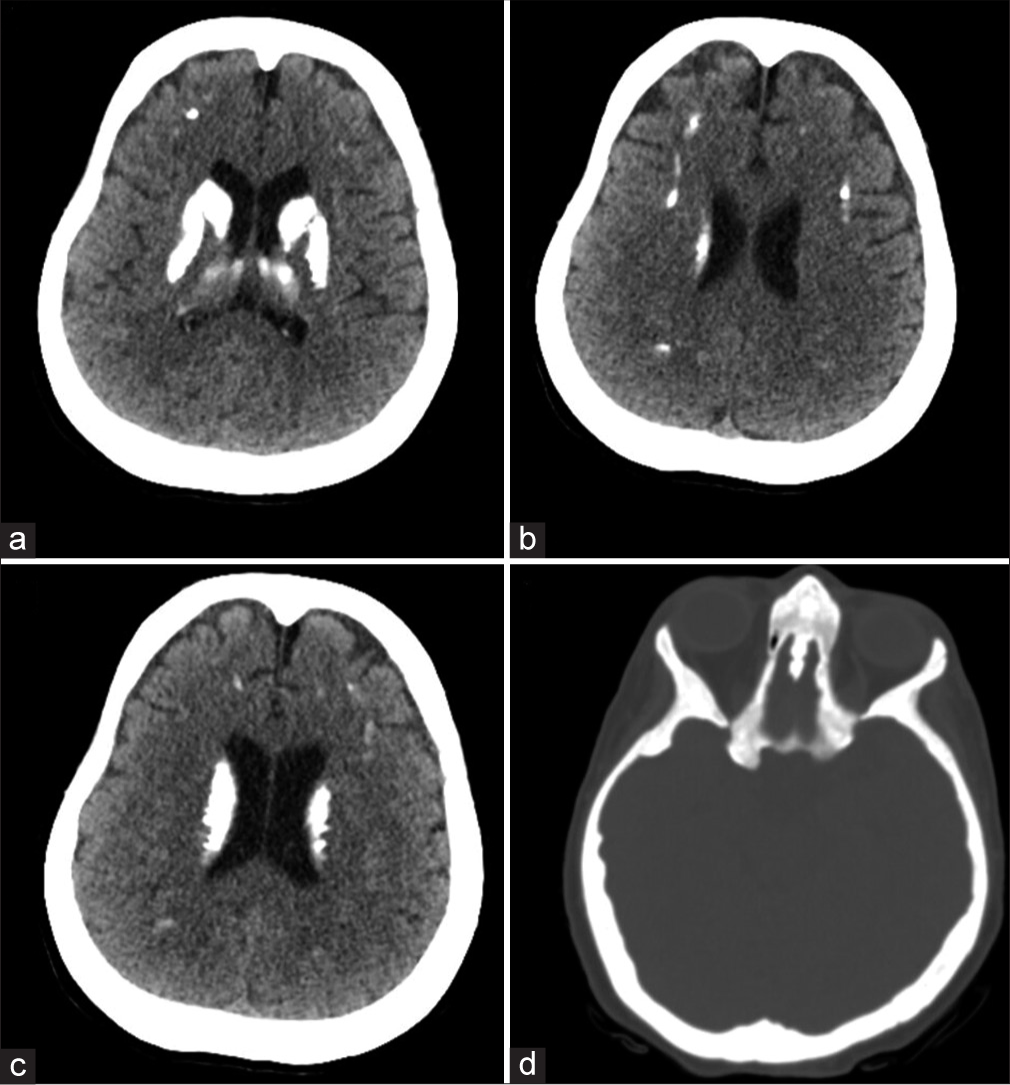
- (a-c) Non-enhanced computed radiography of the brain showing bilateral cerebral coarse calcifications involving basal ganglia, thalami, and periventricular white matter and (d) generalized increased calvarial bone density.
The diagnosis of carbonic anhydrase II deficiency was made as the patient had osteopetrosis, RTA, and cerebral calcification. She was managed with intravenous potassium chloride and was discharged on oral potassium chloride with sodium bicarbonate supplementation. The family refused for genetic studies.
DISCUSSION
Osteopetrosis with RTA and cerebral calcification due to carbonic anhydrase isoenzyme-II (CAII) deficiency is otherwise known as a marble brain disease or Guilbaud– Vainsel syndrome.[1] Patients with CAII deficiency can have other clinical features such as short stature, mental retardation, anemia, retinal disease, and cranial nerve palsies.[2]
CAII is a zinc-containing metalloprotease involved in the hydration of CO2 to form bicarbonate and H+. Normal function of CAII is essential for the renal regulation of acid/base homeostasis and bone reabsorption by osteoclasts. Loss of function of this enzyme causes the combination of RTA with osteopetrosis. CAII is expressed in the proximal and distal part of the nephron causing a mixed type RTA in CAII deficiency.[3]
Most of the patients with CAII deficiency have manifestations due to osteopetrosis and increased bone density, causing fractures of long bones, cranial nerve palsies, and back pain. Our patient presented with limb weakness due to hypokalemia and RTA, which is unusual. Inherited forms of RTA are common in children and adolescents. Mutations in the genes responsible for the secretion of hydrogen ions and bicarbonate reabsorption are responsible for the inherited types of RTA.
RTA secondary to CAII deficiency is usually a combination of the proximal and distal types of RTA. The severity of RTA is also variable in affected individuals.[4] In some patients, the predominant type of RTA can be distal RTA and can result in nephrocalcinosis, as seen in our patient.
Cerebral calcification in CAII deficiency usually develops by 2–5 years of age. The common areas in the brain where the calcifications are seen include the basal ganglia and the cortex. Basal ganglia calcifications primarily appear in the putamen and caudate nucleus. Calcification of the cortex initially involves frontal lobes.[5] Metabolic diseases associated with bilateral basal ganglia calcification include hypoparathyroidism, pseudohypoparathyroidism, and Fahr syndrome. Conditions causing cerebral calcification and RTA include Fahr syndrome and Wilson disease.[6]
CAII deficiency presents late in infancy or early in childhood with fracturing, developmental delay, weakness, short stature, and/or cranial nerve compression and palsy. The skeletal findings may improve by adult life, and CAII deficiency can be associated with a normal lifespan. Diagnosis requires mutational analysis of CAII. At present, there is no established medical treatment for CAII deficiency. Symptoms of metabolic acidosis improve with long-term alkali treatment.[7]
CONCLUSION
The case highlights the importance of considering CAII deficiency in patients presenting with RTA with other features such as osteopetrosis and cerebral calcification.
Ethical approval
Institutional Review Board approval is not required.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
Financial support and sponsorship
Nil.
References
- The Guilbaud-Vainsel syndrome patient presenting with quadriparesis. Turk J Nephrol (Online). 2022;31:78-80.
- [CrossRef] [Google Scholar]
- Autosomal dominant osteopetrosis associated with renal tubular acidosis is due to a CLCN7 mutation. Am J Med Genet A. 2016;170:2988-92.
- [CrossRef] [PubMed] [Google Scholar]
- Carbonic anhydrase II deficiency: A rare case of severe obstructive sleep apnea. Front Pediatr. 2018;6:213.
- [CrossRef] [PubMed] [Google Scholar]
- Renal tubular acidosis, osteopetrosis, and cerebral calcification: A rare syndrome caused by carbonic anhydrase II deficiency. Indian J Nephrol. 2017;27:330-1.
- [CrossRef] [PubMed] [Google Scholar]
- Osteopetrosis with renal tubular acidosis and cerebral calcification. Kidney Int. 2018;93:1020.
- [CrossRef] [PubMed] [Google Scholar]




