
Translate this page into:
Fibrodysplasia Ossificans Progressiva (Stoneman Syndrome) – A Rare Skeletal Dysplasia

*Corresponding author: M. Dhivakar, Department of Radiodiagnosis, Maulana Azad Medical College, Lok Nayak Hospital, Jawaharlal Nehru Marg, Delhi Gate, New Delhi - 110 002, Delhi, India. dhivakarvino@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Dhivakar M, Prakash A, Garg A, Agarwal A. Fibrodysplasia Ossificans Progressiva (Stoneman Syndrome) – A Rare Skeletal Dysplasia. Indian J Musculoskelet Radiol 2020;2(1):69-72.
Abstract
Fibrodysplasia ossificans progressiva (FOP) is an extremely rare skeletal dysplasia with characteristic imaging and clinical findings, which includes bilateral hallux valgus, monophalangic great toes with short and stout first metatarsals, heterotopic ossification of muscles and connective tissues, short broad femoral necks, pseudo exostoses, short and stout first metacarpals, C2-C7 facet joint fusion, large posterior elements, and tall narrow vertebral bodies.
We present a case of an 8-year-old male child who came with complaints of multiple progressive hard swellings over the neck, chest, and abdomen with restriction of movements for a duration of 2 years and deformity of great toe on both sides since birth. On clinical examination, the patient had multiple non-tender hard bony swellings in neck, chest, and abdominal wall with bilateral hallux valgus deformity. Radiographic examination revealed well- defined rib-like ectopic osseous outgrowths in the posterior aspect of neck, soft tissues of chest and abdominal wall, bilateral hallux valgus, monophalangic great toe and short first metatarsals with normal cervical vertebral bodies, posterior elements, short first metacarpals bilaterally, and pseudo exostoses in medial aspect of upper one-third of both tibia. With the above classic findings, the diagnosis of FOP was made. Early diagnosis of the condition is very important in these cases as intramuscular injections, biopsies, and trivial trauma can exacerbate the condition with painful flare-ups.
Keywords
Rare autosomal dominant skeletal dysplasia
Heterotopic soft-tissue ossifications
Characteristic imaging and clinical findings
Trivial trauma
Avoid biopsy

INTRODUCTION
Fibrodysplasia ossificans progressiva (FOP), also known as Stoneman syndrome or Munchmeyer disease, is a rare connective tissue disorder with autosomal dominant inheritance.[1] The diseasecondition is characterized by abnormal ectopic ossification of the tendons, ligaments, skeletal muscles, and other soft tissues of the body.[1] There is no involvement of smooth muscles in this disorder. The reported incidence of this condition is 1 in 2 million people.[1] This disorder has characteristic imaging and clinical findings, which includes bilateral hallux valgus deformity, monophalangic great toes, heterotopic ossification of muscles and connective tissues, short and broad femoral necks, pseudo exostoses, short first metacarpal/metatarsals, C2-C7 facet joint fusion, large posterior elements, and tall narrow vertebral bodies.[2] The ectopic osseous growths have a characteristic pattern of involvement which occurs in craniocaudal, proximodistal, and dorsoventral fashion. The disease is progressive and complicated by restriction of movements at the corresponding sites, respiratory failure, and pulmonary infections. The common cause of death in this condition is due to cardiac and respiratory failure which results due to severe restriction of chest wall movements.[2] As the disorder has characteristic findings, it can be easily diagnosed with plain radiographs alone. Early diagnosis of this disorder is very important to avoid unnecessary invasive investigations like biopsies as even trivial trauma and intramuscular injections exaggerate progression of the disease condition with inflammation.[3] Hence, knowledge of this disease condition is very important for the radiologists to avoid invasive investigations.
CASE REPORT
An 8-year-old male child presented with complaints of multiple hard non-tender swellings over the neck, chest, and abdomen with restriction of movements in neck and back for the duration of 2 years and deformity of great toe on both sides since birth. The patient had no history of pain or previous trauma. There was no history of similar complaints in rest of the family members. On examination, the patient had multiple non-tender hard swellings in neck, chest, and abdominal wall with bilateral hallux valgus deformity. The patient was advised radiographic evaluation.
Radiographs of the neck, chest, and feet were performed. They revealed multiple ectopic osseous growths in soft tissues of posterior aspect of neck, chest, and abdominal wall [Figures 1 and 2], bilateral hallux valgus deformity, monophalangic great toe, and short first metatarsal [Figure 3] with normal cervical vertebral bodies and posterior elements.
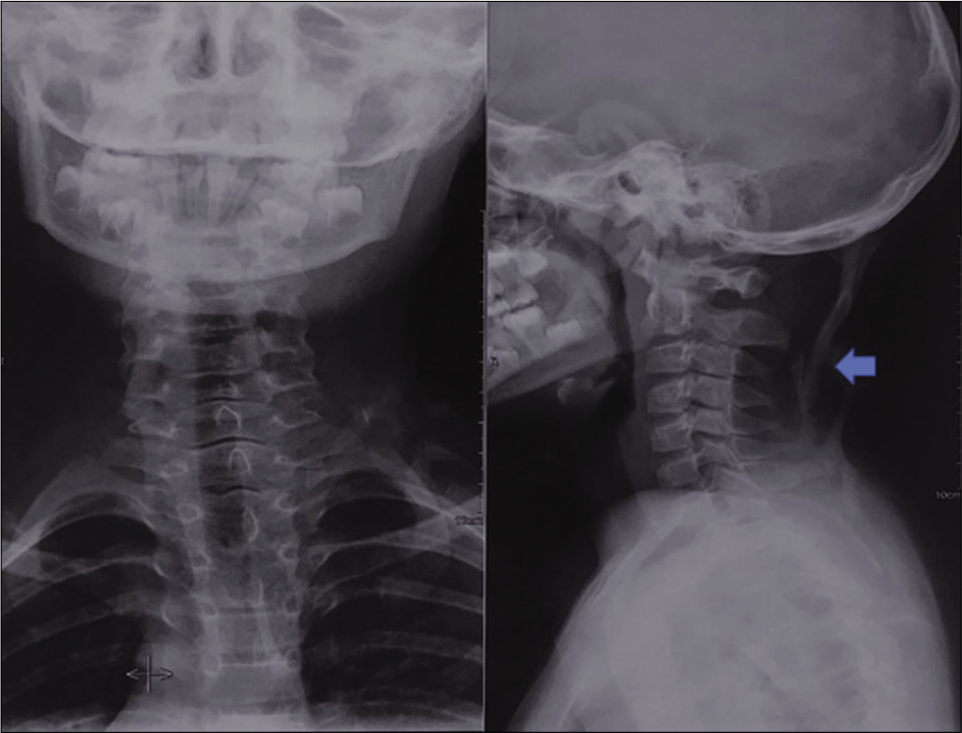
- AP and lateral radiographs of neck reveals heterotopic ossification (blue arrow) in posterior aspect of soft tissues of neck. The cervical vertebral bodies and the posterior elements are normal in this case. The patient had restriction of neck movements.

- Posteroanterior chest radiograph reveals heterotopic ossification (blue arrows) in soft tissues of chest wall and abdomen.

- AP radiograph of both feet reveals lateral deviation of phalanx of bilateral great toes at metatarsophalangeal joint (hallux valgus), single phalanx in bilateral great toe, and short and stout bilateral first metatarsal bones.
With strong suspicion of FOP, the radiographs of chest, abdomen, and foot were followed by the radiographs of hands and knee. These revealed short first metacarpals bilaterally [Figure 4] and sharp bony outgrowths in medial aspect of upper one-third of both tibia [Figure 5] which are called pseudo exostoses due to close resemblance to osteochondromas (exostoses).[1,2] With the above classic findings, the diagnosis of FOP was made. Early diagnosis of the condition is very important in such cases as intramuscular injections, biopsies, and trivial trauma can exacerbate the condition with painful inflammatory flare-ups. No further imaging or invasive investigations were done in this case and the patient’s parents were explained about the condition.

- PA radiograph of bilateral hands show short and stout first metacarpals of both the hands.
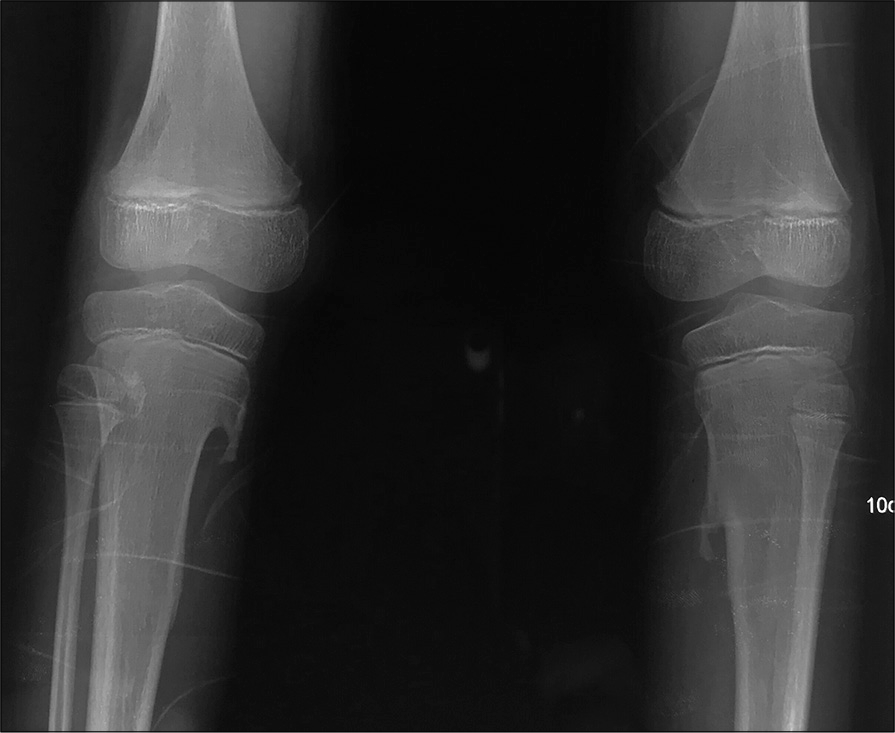
- Pseudo exostoses. AP radiograph of bilateral knees shows sharp bony outgrowths arising from the cortices of upper one- third (metaphysis) of both the tibia which are showing medullary continuity with the tibia.
DISCUSSION
FOP, also called Stoneman syndrome or Munchmeyer disease, is a very rare connective tissue disorder with autosomal dominant inheritance.[1,2] The disorder is characterized by malformation of great toes, thumbs, progressive heterotopic ossification of skeletal muscles, and connective tissue. FOP is an extremely rare disorder with a worldwide prevalence of 1 case in 2 million individuals.[3] It has no racial or gender predisposition. The disorder develops postnatally in the first 10 years of life.
Genetic inheritance pattern is autosomal dominant and can be inherited from either parent. The genetic cause of FOP is due to mutation in activin receptor 1a/activin kinase 2 (ACVR1/ALK2).[4] However, most of the cases arise sporadically as a result of new mutation.[1] It results in ectopic osseous growths in muscles and connective tissues with episodic flare-ups and restriction of body movements at the sites of involvement.
The clinical features of this disorder include progressive ectopic ossification and malformation of great toes. Studies have reported that deformity of the great toe is almost present in all the cases.[5] The great toe malformations such as short metatarsals and hallux valgus were thought to exist from birth and may be a key to an early diagnosis.[6] Patients can also develop acute exacerbations associated with soft-tissue swellings and pain at the affected sites. The heterotopic ossification replaces muscles and connective tissues in the body in a characteristic anatomical pattern. It develops first in proximal, cranial, axial, and dorsal regions of the body and later in distal, caudal, appendicular, and ventral regions of the body.[1] This heterotopic ossification is usually complicated by restriction of movements at the corresponding sites of involvement. It may restrict chest movements leading to an early death due to cardiac and respiratory failure (thoracic insufficiency syndrome).[7] Other commonly associated anomalies include short malformed thumbs, clinodactyly, and proximal medial tibial pseudo exostoses. It can be associated with conductive hearing loss due to ossification of middle ear and weight loss due to ankylosis of jaw.[1]
The characteristic imaging findings include bilateral hallux valgus deformity, monophalangic great toes, heterotopic ossification of muscles and connective tissues, short broad femoral necks, pseudo exostoses, short first metacarpal/ metatarsal, C2-C7 facet joint fusion, large posterior elements, and tall narrow vertebral bodies.[1,2]
The commonly misinterpreted conditions in FOP are aggressive juvenile fibromatosis, dermatomyositis, lymphedema, or soft-tissue sarcoma by the clinicians.[3,5]
Misdiagnosis of FOP is frequent in most of the affected cases mainly because of the hard swelling in the affected regions.[5] However, the characteristic great toe malformation in FOP is not present in these conditions. The affected children often undergo unnecessary and harmful invasive diagnostic biopsies which result in inflammatory exacerbation and progression of the condition. Diagnosis of this condition is mainly based on the clinical and imaging findings. Conventional radiographs alone play a very important role in the diagnosis of the condition. Bone scans are abnormal before conventional radiographs detect heterotopic ossification.[1] Magnetic resonance imaging also plays a role in making early diagnosis before the ossification. The pre-osseous lesions usually have low signal intensity on T1-weighted images and high signal intensity on T2-weighted images.[1,3] Definitive genetic testing of FOP is now available and can confirm diagnosis before appearance of heterotopic ossification.
There is no effective treatment or prevention for FOP.[1] Studies are being done to regulate the overactive ACVR1/ ALK2 signaling pathway that specifically blocks heterotopic ossification.[8] The current management of FOP is mainly supportive and based on early diagnosis of the condition and avoiding injury or iatrogenic harm, symptomatic relief in cases of painful flare-ups, and moderation of residual function. The role of bisphosphonates and corticosteroids during acute exacerbation of the condition is still in research.[1]
All intramuscular injections and invasive biopsies must be avoided as they flare-up the condition with inflammation and faster growths.[1,2,5] Overstretching of jaw during dental procedures causes additional trauma to temporomandibular joint and leads to disease flare-ups.[5] Infections also flare-up the condition.[7] Hence, prophylaxis against influenza and other respiratory tract infections is important. Injuries exacerbate the condition but prevention of trauma is not always possible.[9,10] General anesthesia is particularly dangerous in patients with FOP; hence, it should be avoided.
CONCLUSION
FOP is a rare autosomal dominant connective tissue disorder. Early diagnosis of this condition is very important for genetic counseling, minimizing trauma, and painful flare-ups. Radiological investigations, especially plain radiographs, play a very important role in the diagnosis of this condition by aiding in identification heterotopic ossification, the characteristic great toe changes, and other associated findings. Hence, radiologists must be aware of the diagnostic findings of this condition.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Fibrodysplasia ossificans progressiva-radiological findings: A case report. Oman Med J. 2014;29:368.
- [CrossRef] [PubMed] [Google Scholar]
- Proximal tibial osteochondromas in patients with fibrodysplasia ossificans progressiva. J Bone Joint Surg Am. 2008;90:366.
- [CrossRef] [PubMed] [Google Scholar]
- Fibrodysplasia ossificans progressiva: Report of a case and review of articles. Iran J Radiol. 2011;8:113.
- [Google Scholar]
- Fibrodysplasia ossificans progressiva. Best Pract Res Clin Rheumatol. 2008;22:191-205.
- [CrossRef] [PubMed] [Google Scholar]
- Iatrogenic harm caused by diagnostic errors in fibrodysplasia ossificans progressiva. Pediatrics. 2005;116:e654-61.
- [CrossRef] [PubMed] [Google Scholar]
- Myositis ossificans in children: A review. Eur J Orthop Surg Traumatol. 2017;27:491-502.
- [CrossRef] [PubMed] [Google Scholar]
- Early mortality and cardiorespiratory failure in patients with fibrodysplasia ossificans progressiva. J Bone Joint Surg Am. 2010;92:686.
- [CrossRef] [PubMed] [Google Scholar]
- Disease-causing allele-specific silencing against the ALK2 mutants, R206H and G356D, in fibrodysplasia ossificans progressiva. Gene Ther. 2012;19:781-5.
- [CrossRef] [PubMed] [Google Scholar]
- The fibrodysplasia ossificans progressiva lesion. Clin Rev Bone Miner Metab. 2005;3:195-200.
- [CrossRef] [Google Scholar]
- Deformity of the great toe in fibrodysplasia ossificans progressiva. J Orthop Sci. 2010;15:804-9.
- [CrossRef] [PubMed] [Google Scholar]




